Instructions for running VIBFold
Important: VIBFold is still under construction and being tested. Code is not yet complete, not yet thoroughly tested and devoid of comments.
Instructions
To run VIBFold, three extra files are required: submit_vibfold.py, VIBFold.py and VIBFold_adapted_functions.py. In submit_vibfold.py, you can adapt the following lines in the header of the script to set up your own experiments (note that this is the current state of the script and might be made user-friendly in the future):
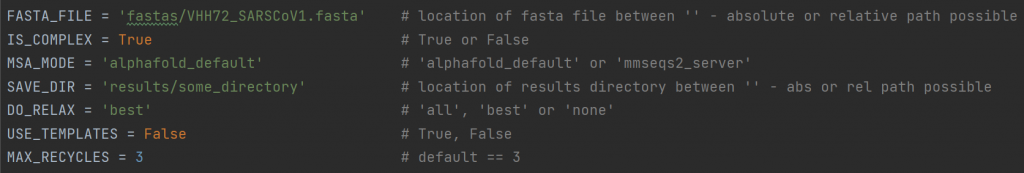
FASTA_FILEIS_COMPLEX) will choose how multiple entries are handled.IS_COMPLEX: For a FASTA file with multiple entries, you can choose whether AlphaFold-Multimer will be run to fold all entries into a protein complex, or whether AlphaFold-Monomer will be run on each entry individually.MSA_MODE: the MSA search can be done with the default AlphaFold MSA search, or via the MMseqs2 serverSAVE_DIR: the location where output directories will be created.DO_RELAX: choose whether you want to have all predictions relaxed, just the top ranked one, or none at all.USE_TEMPLATES: choose whether or not to allow templates used in the predictionsMAX_RECYCLES:(default 3) choose whether or not to include extra recycling steps. We advise to leave this at the default value if you do not know what this means.- EXTRA: In the new version of VIBFold, you can also set the NUM_RUNS_PER_MODEL, indicating how many predictions are made for each of the five AlphaFold models
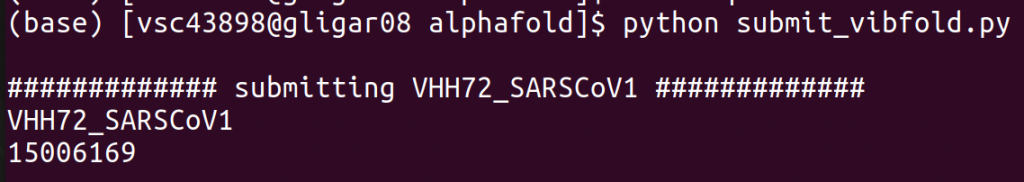
Once you have modified the configuration to your liking, make sure to load the appropriate cluster (e.g., module swap cluster/joltik), and (to be sure) an appropriate python version (module load Python/3.8.6-GCCcore-10.2.0). Finally, run python submit_vibfold.py to start the script.

What happens
A job is automatically submitted for each of the entries in the FASTA (IS_COMPLEX=False) or once for the full FASTA (IS_COMPLEX=True). For each job, a new directory is created in the specified SAVE_DIR. When running a complex, one directory will be created for each permutation of the entries (e.g., A+B and B+A will have separate runs). After the jobs finish, all outputs can be found here.