AlphaFold outputs
After submitting a job script, sit back and relax. Your job is put in a queue on the HPC, and will automatically run when it is your turn to have some resources allocated. After the run is finished, several outputs are returned.
- In the
alphafold/directory, an output and an error file are created, named<job_name>.o<job_id>and<job_name>.o<job_id>(for instance,example_run.o1502930andexample_run.e1502930. Those contain some information about the run, such as the progress made so far, or warnings or errors. If no problems occurred during the job, you can safely remove those files, as the actual AlphaFold output files are located elsewhere. - In the
alphafold/runs/directory, a new directory is created with the FASTA name and the job id of the experiment. In here, the predicted structures and extra information are located.
Alphafold/runs/
In the runs directory, a new directory should be created for the job. Here, a copy of the FASTA file is found, as well as another subdirectory with AlphaFold outputs. Below, a summary is given of their contents. The different file extensions are as follows:
.pdb– protein database format. These files contain the actual structures, including the pLDDT (local confidence) in the b-factor column.pkl– pickle data format, used in programming, but in a binary format an thus not readable from the command line.json– readable data format, often used in programming languages, but contents can be shown withcatand other commands- msa files have
.stoor.a3mfile extensions, readable withcatand other commands
Note that by default, there are no image files attached in the output, for instance visualizing the pairwise PTM scores (global confidence).

The files that take up the most space are usually the MSA (depending on the actual MSA size, but this can take several GBs) and the .pkl data files (grows quadratically with sequence length, with more than 1 GB reached for sequences of more than ~800 amino acids long)
Visualization script
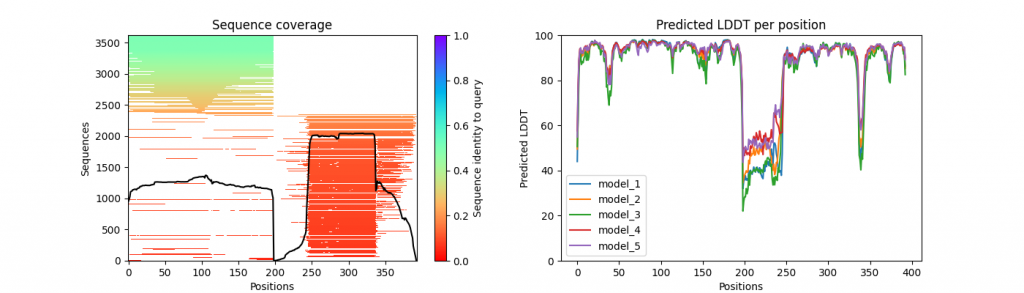
We provide a python script (visualize_alphafold_results.py) to extract pLDDT, PAE and MSA visualizations (inspired by ColabFold code). The script uses the contents of the .pkl output files from the AlphaFold run. It takes three parameters in input:
--input_dirThe location where the AlphaFold output files were stored.--output_dir(optional) The location where the images that are generated should be stored. By default, they are stored in the input directory.--name(optional) The prefix that will be used in for the filenames of the generated files. By default, no prefix is added.
To run the script, you will also have the correct python modules loaded. You can do this by running the following lines before running the actual script.
module load matplotlib/3.7.2-gfbf-2023a
module load AlphaFold/2.3.2-foss-2023a
A full example is shown below:
prefix_coverage_LDDT.png
prefix_PAE.png
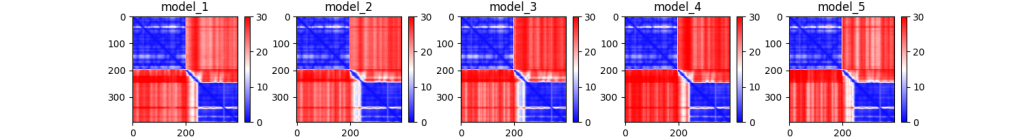
A nice visual tutorial on how to interpret the PAE can be found at the AlphaFold Protein Structure Database website, at any predicted structure, at https://alphafold.ebi.ac.uk/entry/Q9Y223, at the section “Predicted aligned error tutorial”.