Troubleshooting
When attempting to run AlphaFold on the HPC, you might be confronted with a variety of errors, originating from several sources. Most commonly, they will be a result of a wrong configuration or resource exhaustion (e.g., when running large proteins or proteins with too large MSAs). The most common errors are enlisted below, along with possible workarounds to solve them.
You will find the errors that are described in two possible locations:
- At submission time (
qsub), the command line will reply with an error specification - In the error file that is generated for each job running on the HPC. This file can be found in your
$VSC_DATA/alphafolddirectory, and will be named<job_name>.e<job_id>(for instance:example_run.e1502930). Display its contents withlessorcat. The actual error message is most often found at the bottom of the file.
At submission time
Wrong configuration

nodes, ppn, gpus, mem). If you would for instance try to use the key cpu, this is the error that you would get at submission time.
Wrong setup: module not loaded

module swap cluster/joltik), this is the error produced.In the output error file
Wrong setup: wrong FASTA file name
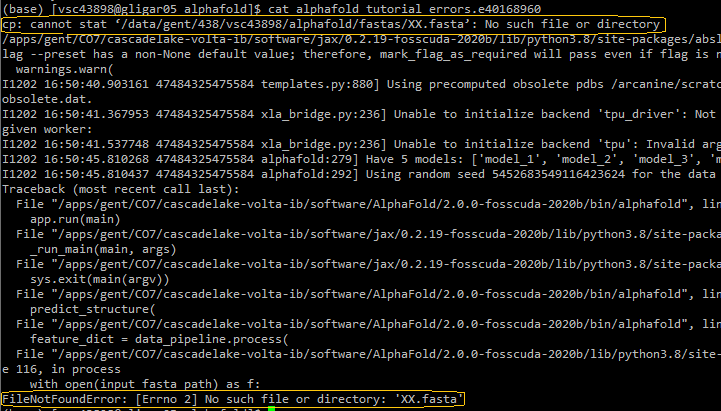
$VSC_DATA/alphafold/fastas/Insufficient CPU memory (RAM)
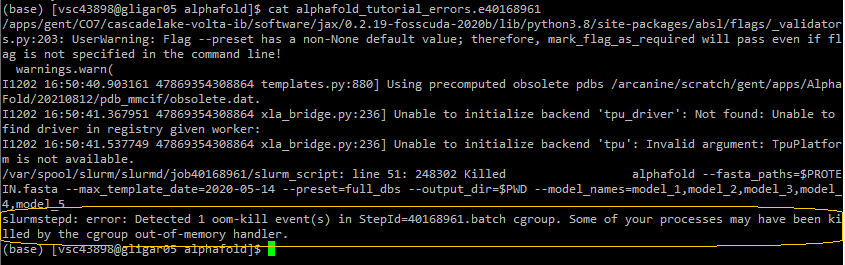
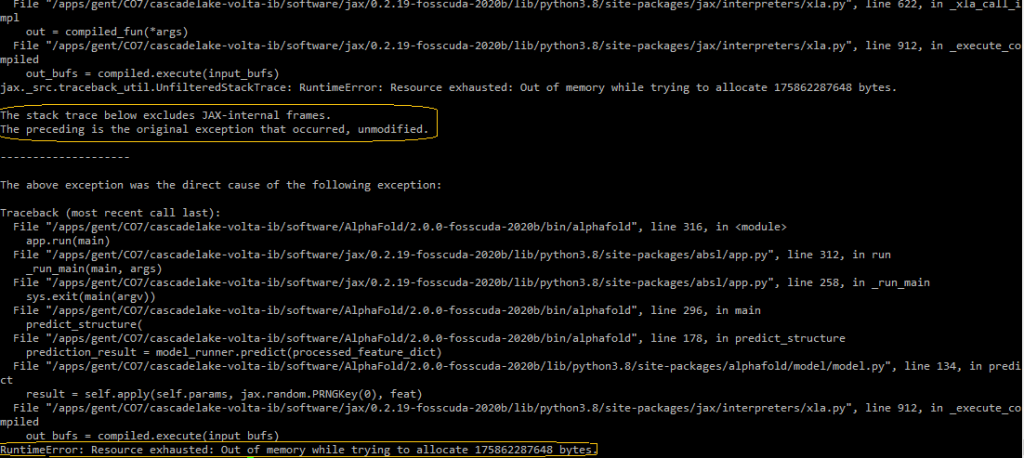
Insufficient GPU memory
Insufficient storage
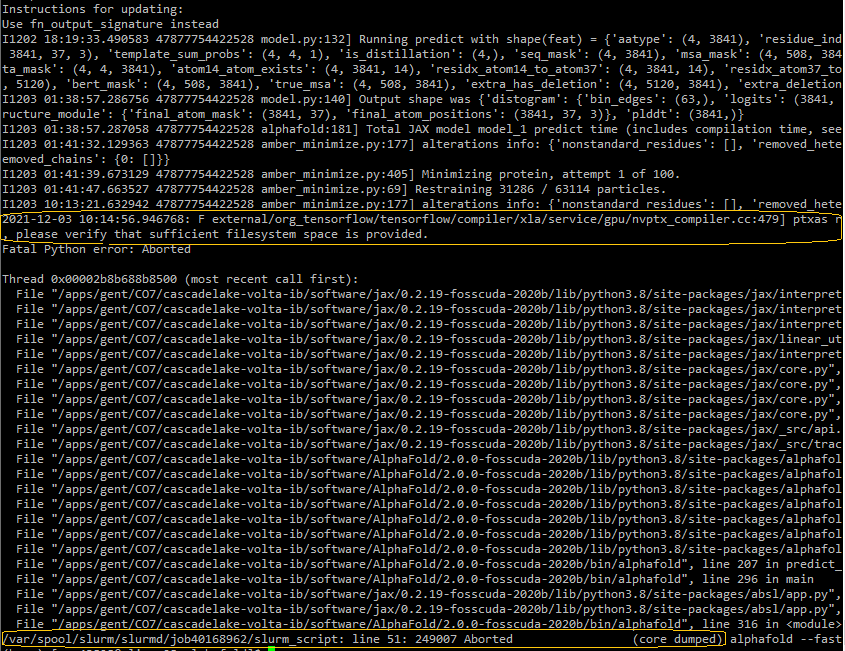
alphafold/ directory is located. If this is in $VSC_DATA, you can try to (a) free up space, or (b) join a virtual organization (VO), where you will have more storage capacity.Time limit exceeded
