AlphaFold training details
To fully interpret AlphaFold predictions, it can be worthwhile to understand the training scheme behind it. Some interesting techniques are used at training time to improve model accuracy and the interpretability of results. Some of those techniques are discussed below.
AlphaFold trains to accurately estimate a local and global confidence for the 3-D structure
One of the most useful tools in the AlphaFold training procedure is the use of several losses. During training, any neural network modifies its parameters with a certain goal in mind. Typically, this goal is represented by a loss function that we want to minimize. In the case of AlphaFold, the loss function mainly tries to minimize the distance between the predicted 3-D structure and the actual PDB structure, which is called the Frame Aligned Point Error (FAPE). However, additional losses are also present:
- Distogram prediction loss
- Masked MSA prediction loss
- Structural violation loss (i.e. in the Structure module, there are no physical constraints imposed on the folding – the models implicitly learns these constraints using this loss)
- …
- Predicted local distance difference test loss (pLDDT)
- Predicted alignment error loss (PAE)
The pLDDTs and PAEs allow us to directly derive a per-position confidence for predicted models. The pLDDT is even stored in the PDB files in output, in the b-factor column. The PAE can be (programatically) found in the .pkl files in output.
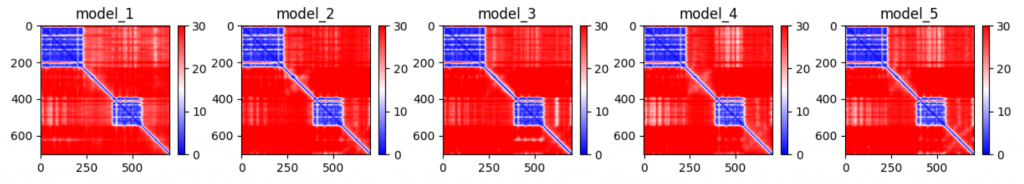
Training is done on protein fragments of max 256/348 residues.
Interestingly, AlphaFold is not trained on full proteins, but rather on protein fragments. In the first part of training, the maximum is set at 256 residues, and later on at 348. This is also the case for AlphaFold-Multimer, where the models are trained to correctly fold interacting proteins. In this case, appropriate subsequences are chosen to include a fair mix of interface and non-interface residues.
Training is done partly on AlphaFold-predicted targets.
Another key part of training is called self-distillation. Here, an initial model is trained on the PDB, after which it predicts 300’000 structures from new sequences. These are then used together with the PDB structures in training the five final AlphaFold models.