(old) Analyze the predicted SARS-CoV-1 + VHH-72 complex (solutions)
Here, you can find an overview of the conclusions that can be drawn from the outputs that were generated by AlphaFold:
1. Model rankings. The overall model confidence is measured in terms of predicted TM score (PTM). These scores per model are listed in the ranking_debug.json file. This file looks as follows:
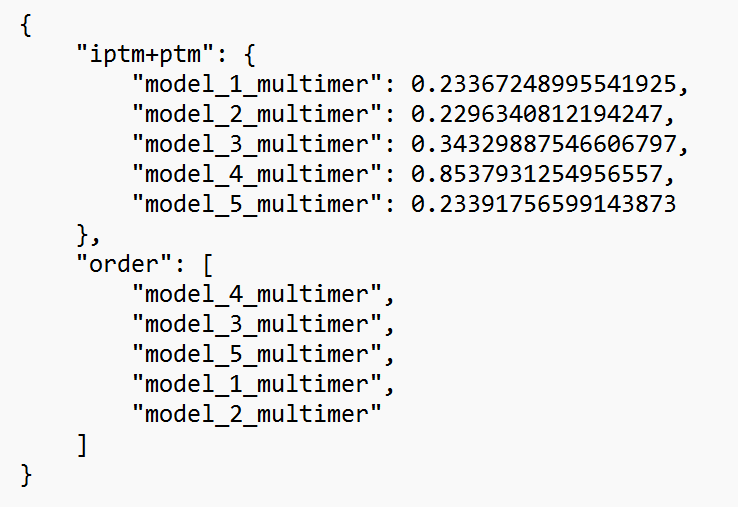
In this file, the PTM score (“iptm+ptm”) is given for each of the five predicted structures. The closer to one, the greater the overall confidence for the model (note: for multimer structures, the weight of the interface region is greater than the other regions). You can see that model #4 is ranked first (in the order part), as it achieves a PTM of 0.85, where other models do not go above a score of 0.34. We can conclude that this fourth model has a high confidence, whereas the other four models are low in confidence.
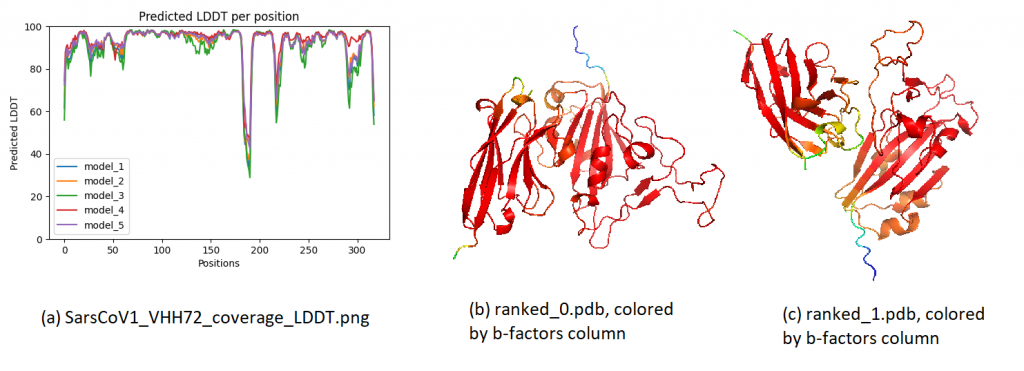
2. PLDDT. The local confidence (per residue) is found by means of PLDDT. These can be visualized in two ways, as shown below. In the visualization, most regions have a very high confidence, meaning that the residues are estimated to fold very accurately locally. This is the case even for the second model (ranked_1), which only achieved a global PTM of 0.34. Some loop/terminal regions have a slightly lower confidence, as often seen.
3. PAE. The predicted aligned error plots gives an idea of how confidently AlphaFold organizes multiple domains or chains relative to each other. Blue blocks are confident, red blocks indicate uncertainty. You can find more information at the “Predicted aligned error tutorial” tab on the AlphaFoldDB.
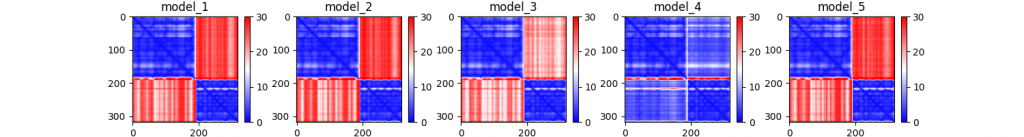
In these visualizations, the fourth model (which ranked first, with a PTM of 0.85) shows the greatest confidence, with both chains aligning very confidently to one another. You can see that the models with much lower PTM scores do not show this characteristic (because of the red blocks). This means that AlphaFold does not predict the interaction properly, despite predicting the chains with high accuracy.
4. Aligning predicted structures to 6WAQ.
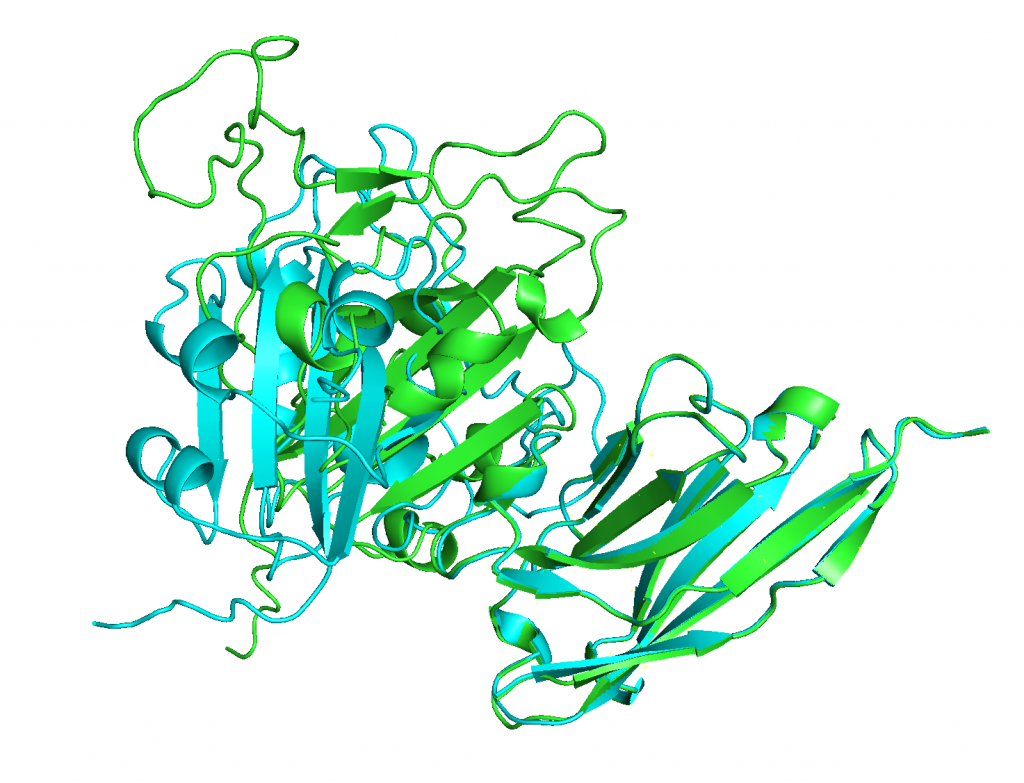
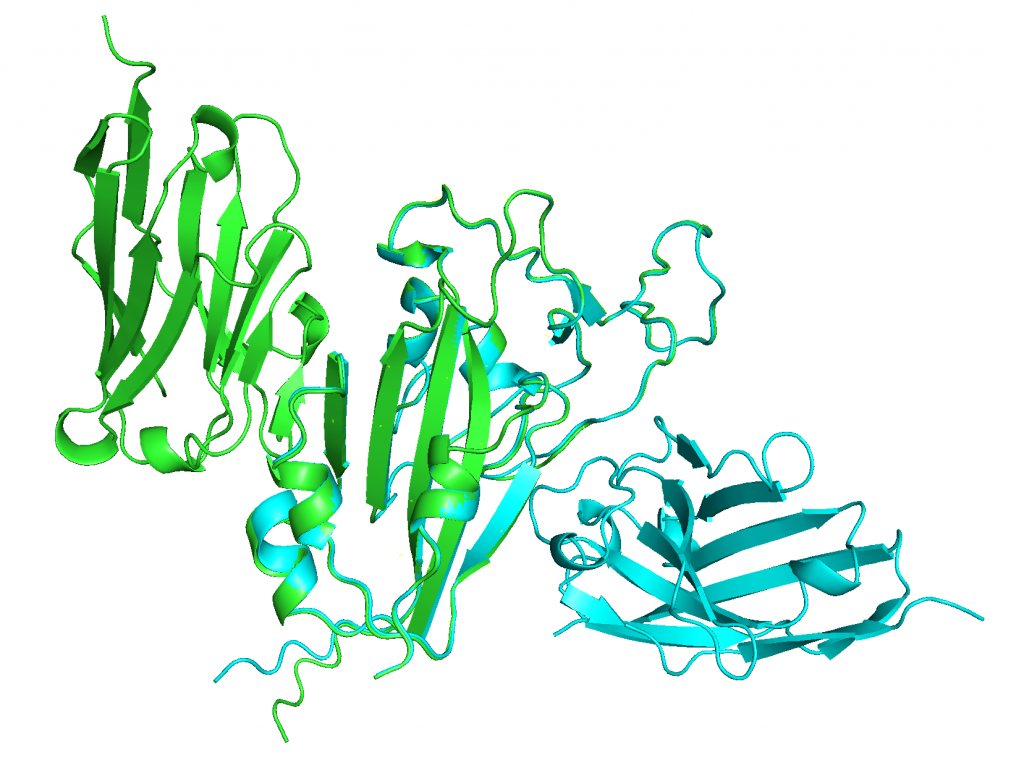
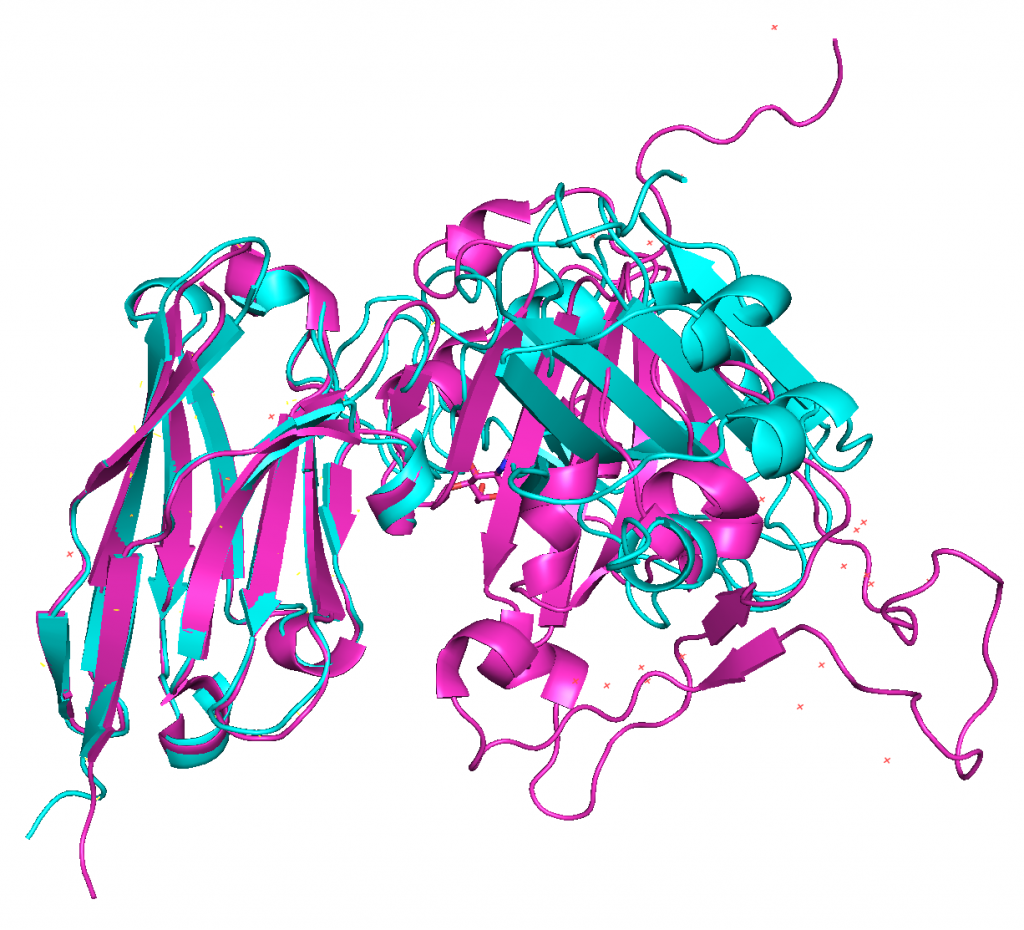
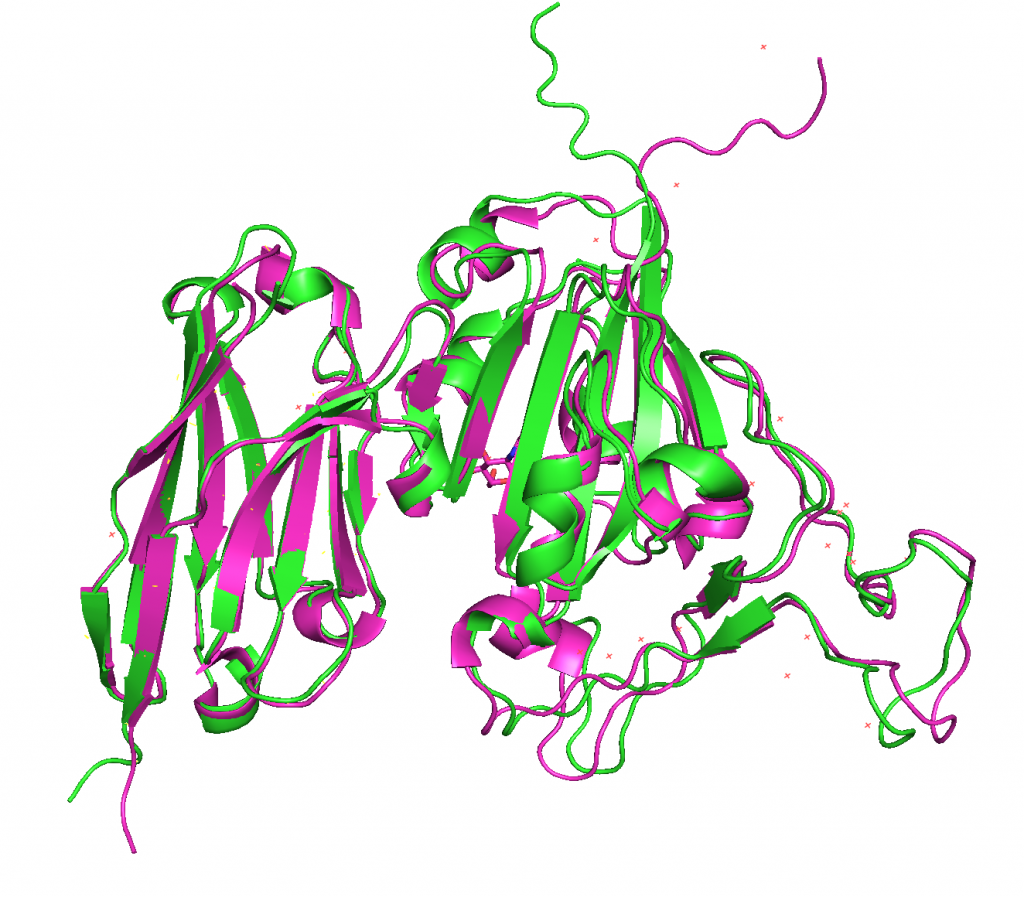
ranked_0.pdb on the other hand does align nicely with 6WAQ.