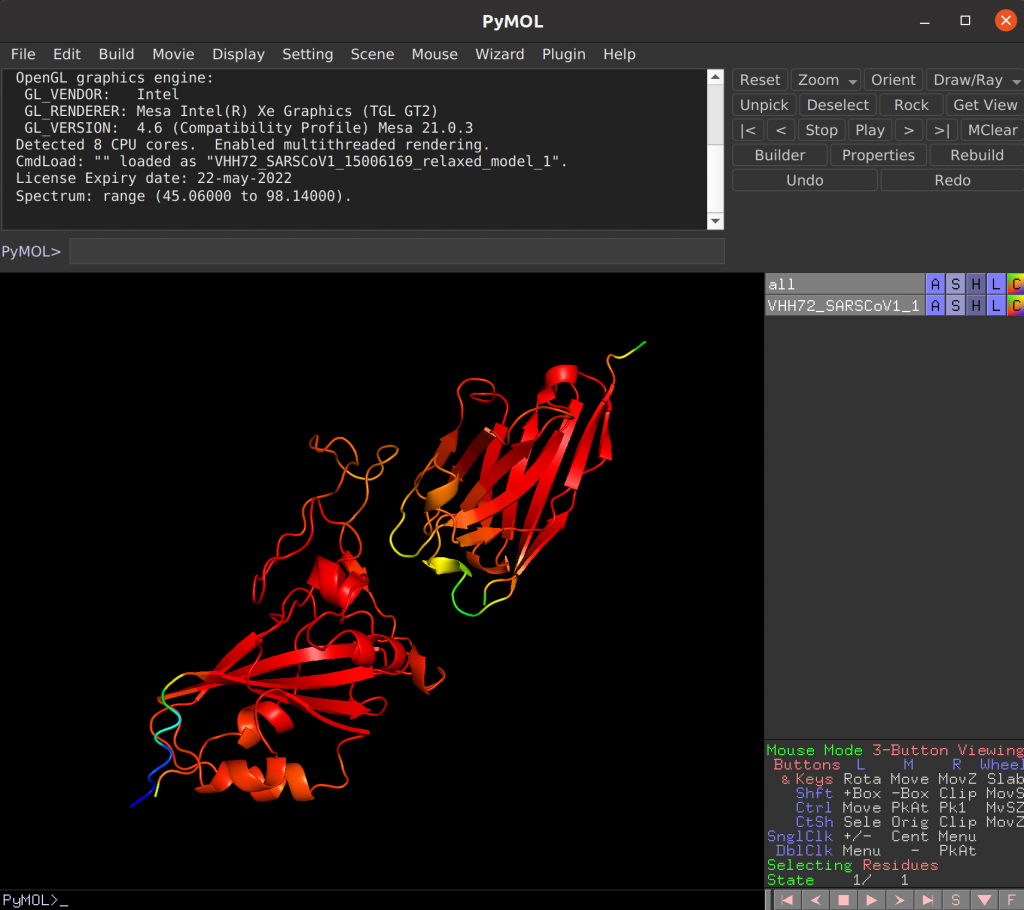
Overview of VIBFold outputs
Outputs for AlphaFold-Multimer or the default AlphaFold-Monomer runs are the same. An example for the Multimer model is given below.
File naming. The protein ids (monomer) or fasta file name (multimer) are used at the start of the directory name, followed by the job id, and for the directory name: in case of multimer, by the number of the permutation.
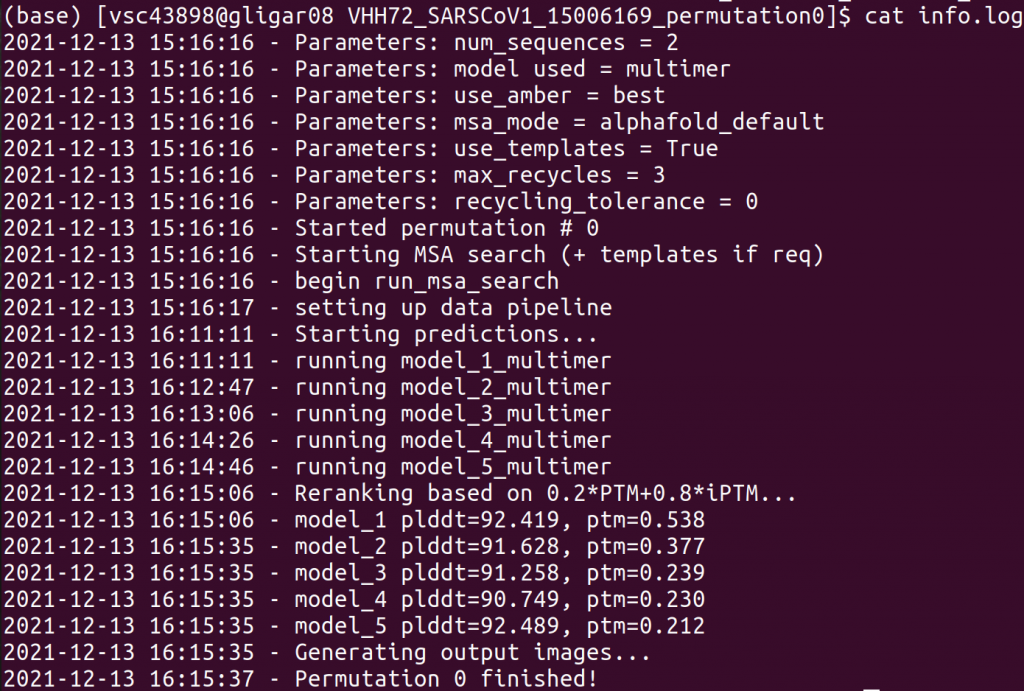
info.log
This information file provides several forms of information:
- an overview of the parameters for this particular run
- timing information for how much time each step required
- ranking information: on what ranking is based and what the scores are for each prediction
query.fasta
The FASTA file that was used for this run.
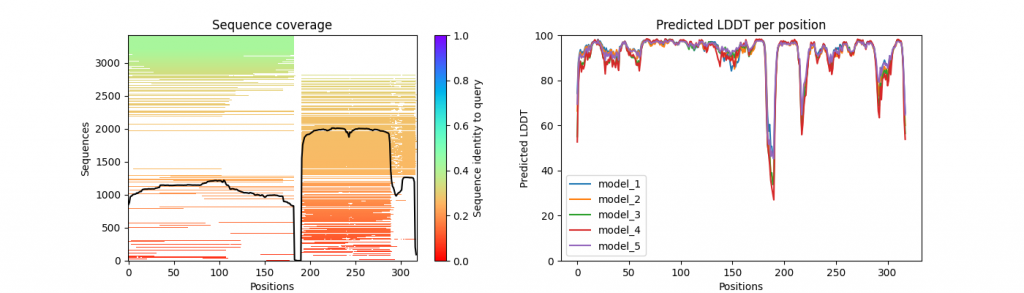
…_coverage_LDDT.png
An illustration of the MSA coverage and sequence similarity, together with the pLDDT per position for each of the five models.
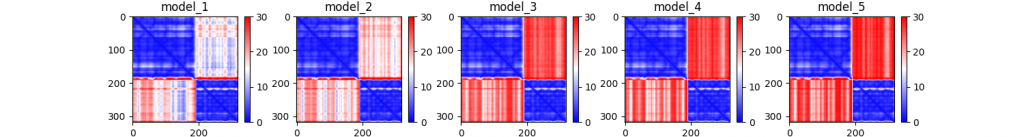
…_PAE.png
An illustration of the predicted aligned error for each of the five models.
…_unrelaxed_model_X.pdb
The unrelaxed .pdb structures for each prediction model. Note that the b-factor column also holds the pLDDT per position.
…_relaxed_model_X.pdb
Depending on the configuration, the best model, all models or none of the models will be relaxed using AMBER. The results can be found in the _relaxed_X.pdb files. Note that the b-factor column also holds the pLDDT per position.